第二节 分子生物学实验基础
分子生物学是在生物化学基础上发展起来的,以研究核酸和蛋白质结构、功能等生命本质的学科,在核酸、蛋白质分子水平研究发病、诊断、治疗和预后的机制。其中基因工程(基因技术,基因重组)是目前分子生物学研究热点,这些技术可以改造或扩增基因和基因产物,使微量的研究对象达到分析水平,是研究基因调控和表达的方法,也是分子水平研究疾病发生机制、基因诊断和基因治疗的方法。转化(transformation)、转染、转导、转位等是自然界基因重组存在的方式,也是人工基因重组常采用的手段。基因重组的目的之一是基因克隆(gene clone),基因克隆可理解为以一分子基因为模板扩增得到的与模板分子结构完全相同的基因。使需要分析研究的微量、混杂的目的基因易于纯化,得以增量,便于分析。
外来基因引起细胞生物性状改变的过程叫转化(transformation),以噬菌体把外源基因导入细菌的过程叫转染(transfection)。利用载体(噬菌体或病毒)把遗传物质从一种宿主传给另一种宿主的过程叫转导(transduction)。一个或一组基因从一处转移到基因组另一处的过程叫转位(transposition),这些游动的基因叫转位子。
一、基因工程的常用工具
(一)载体
载体(Vector)是把外源DNA(目的基因)导入宿主细胞,使之传代、扩增、表达的工具。载体有质粒(plasmid)、噬菌体、单链丝状噬菌体和粘性末端质粒(粘粒)、病毒等。载体具有能自我复制;有可选择的,便于筛选、鉴定的遗传标记;有供外源DNA插入的位点;本身体积小等特征。
质粒存在于多种细菌,是染色体(核)以外的独立遗传因子,由双链环状DNA组成,几乎完全裸露,很少有蛋白质结合。质粒有严紧型和松弛型之分。严紧型由DNA多聚酶Ⅲ复制,一个细胞可复制1-5个质粒。而松弛型由DNA多聚酶Ⅰ复制,一个细胞可复制30-50个质粒,如果用氯霉素可阻止蛋白质合成,使质粒有效利用原料,复制更多的质粒。质粒经过改造品种繁多,常用的有pBR322、pUC系列等。这些质粒都含有多个基本基因,如复制起动区(复制原点Ori),便于复制扩增;抗抗生素标记(抗氨芐青霉素Ap[SB]r[/SB]、抗四环素Tc[SB]r[/SB]等)或大肠埃希菌部分乳糖操纵子(E.coli LacZ)等,便于基因重组体的筛选;基因发动子(乳糖操纵子Lac、色氨酸操纵子Trp等)和转录终止序列,便于插入的外源基因转录、翻译表达。质粒上还有许多限制性内切酶的切点,即基因插入位点,又叫基因重组位点,基因克隆位点。
常用噬菌体载体有单链噬菌体M13系统;双链噬菌体系统。噬菌体应和相应的宿主细胞配合使用。以上载体各有特点,便于选择,灵活应用。
(二)工具酶
工具酶是基因重组技术不可缺少的工具。主要有限制性内切酶、连接酶、核酸聚合酶、逆转录酶、核酸酶等。
限制性内切酶有Ⅰ型和Ⅱ型限制性DNA内切酶之分,Ⅱ型能严格识别核酸序列,并在识别区内特定的核苷酸处切开DNA双链。故通常所指都是Ⅱ型限制性DNA内切酶。识别分四核苷酸和六核苷酸,其序列旋转对称。切口分开端和粘端,产生3′-OH和5′-P末端。内切酶品种多,使用时应注意温度、缓冲液用量(一般1μg DNA/2-5单位酶)等反应条件。
| 酶 | 识别序列 | 切口 | |
| Alu Ⅰ | …AGCT… | …AG CT… | 四核苷酸 |
| …TCGA… | …TC GA… | 平端切口 | |
| Eco R1 | …GAATTC… | …G AATTC… | 六核苷酸 |
| …CTTAAG… | …CTTAA G… | 粘端切口 |
连接酶有T4噬菌体DNA连接酶、T4噬菌体RNA连接酶、大肠埃希菌DNA连接酶等。DNA连接酶可连接平端,也连接粘端。反应需有Mg[SB]2+[/SB]和ATP存在,pH7.5-7.6。最适温度37℃,30℃以下活性明显下降,但考虑到被连接DNa 的稳定性和粘性末端的退火温度,一般平端连接用20-25℃,粘端连接用12℃左右。
聚合酶有DNA聚合酶(以DNA为模板合成DNA大肠埃希菌DNA聚合酶Ⅰ,大肠埃希菌DNA聚合酶Ⅰ大片段(Klenow大片段),T4或T7噬菌体DNA聚合酶等);RNA聚合酶(以DNA为模板合成RNA,T7或T3噬菌体RNA聚合酶);逆转录酶(以RNA为模板合成DNA,除RNA病毒中发现外,发现大肠埃希菌DNA聚合酶Ⅰ和Taq DNA聚合酶都有逆转录活性)。
大肠埃希菌DNA聚合酶Ⅰ具有5′→3′聚合酶活性和5′→3′,3′→5′外切酶活性。Klenow片段是DNA聚合酶Ⅰ被枯草杆菌酶作用产生的一个大片段,有5′→3′聚合酶和5′→3′外切酶活性,无3′→5′外切酶活性。可用于缺口翻译(Nick translation)法标记核酸,也可用于DNA序列测定,修补DNA链等。
核酸酶有DNase、RNase、核酸酶S1等,可水解相应的DNA和RNA,核酸酶S1可降解单链DNA和RNA,用量增大也可降解双链核酸。它可用于切去ds-cDNA合成中产生的发夹环。
末端转移酶在Mg[SB]2+[/SB]存在下,选择3′-OH端单链DNA为引物加成核苷酸,在Co[SB]2+[/SB]存在下,选择3′-OH端双链DNA为引物加成核苷酸,形成多聚核苷酸尾。常用于核酸末端标记和核酸连接的互补多聚尾(连接器)。
碱性磷酸酶去除5′-P,可防止二分子DNA片段5′端P基团自身空间障碍,影响DNA分子之间的连接,一般用碱性磷酸酶处理载体DNA除去5′端P基团,在连接酶作用下目的基因的5′端P先与载体3′端OH连接,再通过复制修复另一条链,使二条链完全连接。该方法大大提高了连接效率(图18-1)。
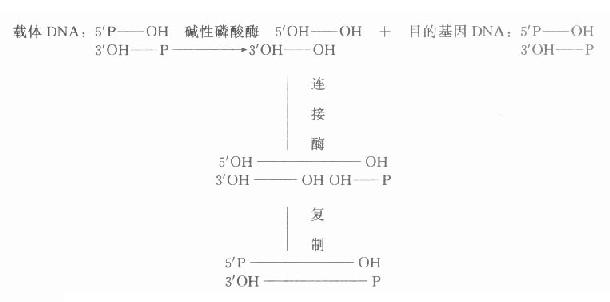
图18-1 经碱性磷酸酶处理后载体DNA与目的基因DNA的连接
二、目的基因
常把需研究的基因称为目的基因,需分析的基因称靶基因,在基因克隆过程中有时两者均称为插入基因,有时三者含义相近。简单的原核生物目的基因可从细胞核中直接分离得到,但人类的基因分布在23对染色体上,较难从直接法得到。简短的目的基因可在了解一级结构或通过了解多肽链一级结构氨基酸编码的核苷酸序列基础上人工合成。但多数的目的基因由mRNA合成cDNA(complementaryDNA,反转录DNA)得到。CDNA通过各种方式与载体连接,克隆可得到全长cDNA或片段,用于探针制备、序列分析、基因表达等研究。因此以cDNA为研究材料反映了mRNA的转录及对以后翻译的影响情况,即反映某一基因(DNA)外显子的情况。(图18-2)
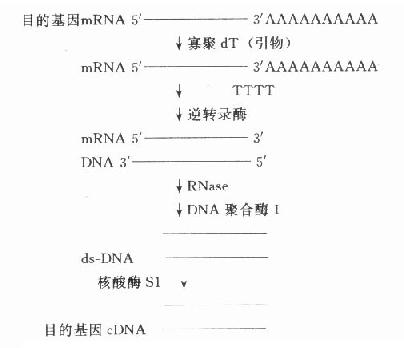
图18-2 目的基因cDNA的合成
三、基因库的建立
建立基因库的目的是为了便于目的基因的保存、扩增和纯化。基因库是利用工具酶,通过基因重组技术和转化、筛选而建立,也是基因克隆的过程。实际上是利用低等生物如细菌、病毒、噬菌体等生长快,繁殖力强的特点,而作为一种核酸扩增筛选的载体,把人类等高等生物的基因或其片段用工具酶插入,重组于其中,经筛选,克隆得到目的基因与载体重组的重组基因,成为便于保存,取用方便的基因库。基因库交流是研究室之间交流目的基因的常用方式。
(一)基因重组
基因重组方案很多,简单的可用同一种限制性内切酶分别在载体和目的基因切出相对应的切口,便于连接。也可用末端连接酶合成连接器(图18-3)。近年,Okayama方案为实验室普遍采用,该方法可得到全长的cDNA。
(二)转化
转化目的是把有复制能力,但在细胞外无复制活性的目的基因-载体重组体装入细胞(大肠埃希菌),使目的基因随载体在细胞内复制、扩增。实验步骤介绍如下:
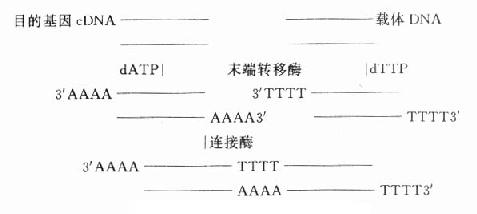
图18-3 基因重组方案之一
⒈大肠埃希菌的处理(增加细胞膜通透性,便于基因进入细胞)大肠埃希菌(E.Coli cells)接种于Lb agar培养基,37℃培养一晚。挑选3~5个大菌落,接种于50ml LB培养基,37℃,振摇培养一晚后,在A[XB]550[/XB]测定,要求细菌繁殖一定量(一般为细菌对数生长期),野生型(rec+,5x10[SB]7[/SB]cells/ml)为0.2-0.3,缺陷型(rec-,5x10[SB]7[/SB]cells/ml)为0.5-0.6。离心弃去上清液,用20ml,50mmol/L,CaCl[XB]2[/XB]悬浮菌体。冰浴20分钟,低温离心。弃上清液,用2.5ml冰50mmol/l CaCl[XB]2[/XB]悬浮。4℃可保存48小时,用于转化,称competent cells。
⒉质粒(如经基因重组建立的重组质粒,基因库等)的转化 将competent cells(以美国BRL公司DH10B菌株为例)置冰水浴。同时将Falcon2059(传热系数稳定)塑料试管置冰水浴。取100μl菌液(DH10B)于2059试管中,加10倍稀释的巯基乙醇1μl,冰浴轻摇2分钟,放置10分钟。取0.1-50ng质粒加入试管,轻轻摇匀,冰浴30分钟。此间备好42℃水浴。并将SOC培养基置42℃备用。将试管置于42℃,轻摇45秒钟,马上置冰浴2分钟。使competent cells热胀冷缩,把质粒导入菌体。加42℃SOC培养基0.9ml于试管,37℃培养1小时。1000rpm离心10分钟,弃上清液,用200μlSOC培养基悬浮菌体。接种于LB-氨芐青霉素(100U/ml)培养皿,37℃培养一晚。已转化的菌体可形成菌落(因质粒上有抗氨芐青霉素基因)。在了解目的基因或其产物的基础上对菌落进行鉴定。并在LB培养液中扩大培养。基因库的建立、转化、筛选、扩增、纯化的过程就是基因克隆的过程。
LB培养基(1L):10g蛋白胨,5g酵母膏,10gNaCl,高压灭菌,pH7.5。
SOB培养基(1L):20g蛋白胨,5g酵母膏,0.5gNaCl,高压灭菌。另外准备2mol/L镁溶液(1mol/L氯化镁与1mol/L硫酸镁等量混匀,过滤灭菌),临用前加1ml于100ml培养基。
SOC培养基(100ml):临用前加入1ml过滤灭菌的2mol/L葡萄糖。
四、核酸的分离与纯化
核酸存在于多种细胞,如病毒、细菌、寄生虫、动植物细胞;多种标本中,如血液、组织、唾液、尿液等其它来源的标本。因此分离方法复杂而多样。又因各种DNA、RNA的丰度差异,各种分析方法对核酸的纯度与量的要求有差异,因此在实验前应对采用的方法有所了解和选择。总的说来核酸的分离与纯化是在溶解细胞的基础上,利用苯酚等有机溶剂抽提(核酸溶于缓冲液,即水相),分离,纯化;乙醇、丙酮等有机溶剂沉淀,收获。溶解细胞的方法因标本不同而不同,有用SDS加NaOH,有用蛋白酶,有的用超声波破碎等方法,苯酚提取主要使蛋白质变性沉淀于有机相,而核酸保留在水相,达到分离核酸的目的。实验上生物标本中含量最多的就是蛋白质。为了除去分离过程中残留的有机溶剂,常用的方法是加冷乙醇和盐沉淀核酸,通过离心回收核酸,然后用70%-80%乙醇洗涤沉淀,除去多余的盐,以免影响核酸溶解和抑制后续步骤的酶促反应。为了得到纯的核酸可用蛋白酶除去蛋白,用RNA酶除去RNA,得到纯的DNA,用DNA酶除去DNA而获得RNA。目前开发了许多商品化的核酸分离柱,可简单、快速地分离得到纯度很高的DNA或RNA。其分离原理有的利用核酸的分子量差异,有的利用需分离核酸的特点与其特异性结合达到分离、回收的目的。
(一)质粒的分离与纯化
含质粒的E. Coli cells经LB培养液250ml扩大培养,倒入50ml离心管,4℃,3000rpm,离心15分钟。弃去上清液。(弃去液丢弃前应作消毒处理,以免污染环境)。加lysis buffer(50mmol/L Glucose,10mmol/l EDTA,25mmol/l Tris-HCl,pH8.0)1-2ml使之悬浮。放置室温5分钟,加3.5-7ml新配制SDS/NaOH溶液(1mol/l NaOH 8ml,20%SDs2ml,蒸馏水30ml)上下摇匀,置冰水浴5分钟。禁用混匀器,以免DNA分子断裂),加2.5mol/L醋酸钾缓冲液(pH4.8)2.6-5.2ml,上下摇匀,冰浴5分钟。4℃,3000rpm,离心15分钟。沉淀蛋白质。取上清液,加无水乙醇12-24ml(上清液的二倍)放室温15分钟后,10000rpm离心,弃上清液。加约为沉淀物体积的0.4倍TE(10mmol/lTris-Hcl pH8.0,10mmol/l EDTA)溶解沉淀后,加0.2倍的7.5mol/L醋酸铵。可用混匀器混匀,置冰浴10分钟。4℃,10000rpm离心5min,弃上清液。加沉淀物0.4倍的10mmol/lTris-HCl pH7.5,10mmol/l EDTA缓冲液,1/10体积的5mg/ml RNase,37℃,30分钟保温。加等量的phenol(酚)/CIAA(480ml氯仿,20ml异戊醇),混匀。4℃,10000rpm,离心5分钟。取上层液加酚/CIAA重复一次。取上层液加1/10体积5mol/l NaCl和2倍无水乙醇,―20℃过夜或―70℃2小时以上。4℃,10000rpm,离心10分钟,弃上清液。加80%乙醇不摇动,直接10000rpm离心,弃上清液,真空干燥。加适量(约200μl)TE溶解。测定含量后备用。
(二)重组质粒中目的基因的分离与纯化
先取少量纯化的重组质粒,用限制性内切酶切出目的基因,经电泳分离,确认。以200μl含2mg DNA(重组质粒)为例:取10μl含100μg DNA,加90μl TE。按1μgDNA加2-5U限制性内切酶,1/10体积缓冲液,1-2小时保温。缓冲液种类和酶反应温度因内切酶种类而异。1/10体积3mol/l NaAc,2倍无水乙醇,-70℃,2小时(-20℃过夜),10000rpm,10分钟离心,弃上清液(乙醇沉淀)。适量TE溶解沉淀(切断的DNA质粒与目的基因混合物),电泳分离(用相应分子量DNA标准同时电泳),在紫外光下观察结果,与标准DNA分子量比较,确认目的基因和内切酶的切割效果。
⒈电泳条件:
| 凝胶制备: | 1%琼脂糖凝胶 | agarose(mg) | X50TAE(ml) | EB(溴化乙锭μl) |
| (agarose) | 1000 | 2.0 | 4.0 | |
| 总体积(ml) | ||||
| 100 |
| 胶浓度(%): | 0.3 | 0.6 | 0.7 | 0.9 | 1.2 | 1.5 | 2.0 |
| DNA长度(kb): | 60-5 | 20-1 | 10-0.8 | 7-0.5 | 6-0.4 | 4-0.2 | 3-0.1 |
| 电泳缓冲液: | X50TAE(ml) | EB(μl) | 总体积(ml) | ||||
| 20 | 30 | 1000 |
X50TAE:Trismabase 54g;乙酸57.1ml;0.5m EDTApH8.0 20ml;蒸馏水至1000ml。
EB:10mg/mlethidium bromide。(EB有致癌性,操作应小心)。
经电泳确认后,取适量DNA(重组质粒)同上条件切开,用1.5%低熔点(<65℃熔解)琼脂糖凝胶,电泳分离。在紫外光下,切出含目的基因区带(凝胶),放入离心管中,提取纯化。
⒉从低熔点凝胶提取,纯化DNA片段 加与凝胶体积相等的TE(10mmol/lTris-HCl pH8.0,0.1mmol/l EDTA),置65℃水浴5分钟保温,使凝胶完全溶解。待放至室温,加等量酚(TE饱和,TE封在上层,取下层酚),轻轻混匀(不用混匀),12000rpm,3分钟离心。反复1-2次。取上层液,加0.1体积3mol/L醋酸钠(pH5.2)和2.5倍体积无水乙醇,进行乙醇沉淀。将纯化的DNA加适量TE溶解,测定含量,备用(可用于目的基因结构分析,探针制备等)。除低熔点凝胶回收法外,如用一般凝胶可用透析带短暂电泳,离心管低部加玻璃棉,高速离心等方法回收DNA片段。
(三)标本DNA的分离与纯化
用5-10ml 1xNTE(NaCl 100mmol/L ,Tris-HCl10mmol/l pH7.4,EDTa 1mmol/L pH8.0)调整细胞为2×10[SB]7[/SB]个(组织应切碎置液氮冰冻,高速搅切成粉末后,加入缓冲液)。加1/10-1/20体积(V)10mg/ml蛋白酶(Sigma Ⅷ型),1/20v 10%SDS,37℃2小时。加等量酚/3xNTE(3xNTE上封)混匀(至少7分钟)。4℃,3000rpm,10分钟。取上清液,加2mlTE(Tris-HCl 10mmol/L pH7.5,EDTa 1mmol/L pH8.0),充分混匀。乙醇沉淀。2mlTE溶解沉淀。加1/30xNTE,蛋白酶K(10mg/ml)代替蛋白酶重复2-4步骤。测定含量。
(四)样品RNA的分离,纯化
含10[SB]6[/SB]个细胞液加等量纯化溶液[4mol/L胍基硫氰酸盐,25mmol/L柠檬酸pH7.0,0.5%肌氨酸(sarcosyl),0.1mol/l 2-巯基乙醇]。总体积为细胞沉淀4-5倍,混匀。加0.1体积2ml/L乙酸钠(pH4.1),1体积酚(重蒸水上封),0.2体积CIAA,混匀器剧烈混合10秒钟。冰水浴15分钟。10000xg4℃,20分钟。离心(不用刹车)。小心取出上清液。加等量丙酮(或2倍乙醇),-20℃,60分钟以上。10000xg,4℃,20分钟离心。弃上清液。用1.5ml离心管约加0.3ml纯化溶液,重复3)步骤,离心10分钟,弃上清液。加75%乙醇,10000xg,4℃,10分钟离心。弃上清液。真空干燥15分钟,备用。
五、探针制备
探针制备就是目的基因的标记,核酸标记方法常用的有缺口平移法、随机引物法、末端标记法等。标记物质有放射性元素(如[SB]32[/SB]P等)和非放射性物质(如生物素、地高辛等)。[SB]32[/SB]P是最常用的核苷酸标记同位素,被标记的dNTP本身就带有磷酸基团,便于标记,特点是比活性高,可达9000Ci/mmol;发射的β射线能量高,可达1.70MeV,用它标记的探针自显影时间短,灵敏度高。[SB]32[/SB]P的半寿期为14天,应随标随用,一般标记后,在一周内使用,它虽带来不便,但给使用后废弃物处理减轻了压力。使用[SB]32[/SB]P标记物应注意防护,操作时应有1-1.5cm厚聚甲基丙烯酸甲酯有机玻璃隔离保护,避免直接照射。操作人员胸前应佩带个人计量器,定期检测。结束实验后,应用专用探测器(盖革-米勒检测器),检查工作区域、手、衣服等,以免污染发生。其它同位素标记物,同样应注意放射线防护。
非放射性标记有酶标和化学物标记法。酶标方法与免疫测定ELISA方法相似,只是被标记的核酸代替了被标记的抗体,事实上被标记的抗体也称为探针,阅读文献时应加以注意。现有许多商品是生物素(biotin)、地高辛标记的,如生物素-dUTP、生物素-dATP、地高辛-dUTP等。血凝素(avidin)与生物素有非常高的亲和性,当血凝素标记上过氧化物酶或碱性磷酸酶(血凝素-酶),经杂交反应最终形成:探针-生物素-血凝素-酶复合物(ABC法)。酶催化底物显色,观察结果。与一般的酶反应底物不同,ABC法底物显色后为不溶物,以便观测结果。酶标记法复杂、重复性差、成本高,但便于运输保存,灵敏度与放射物标记相当。化学物标记有的也是利用相应酶标抗体形成特异复合物,与上述方法相当,有的则可自发光。化学标记法简单、成本低,但灵敏度相对较低。(表18-1)
表18-1 探针标记及特性
| 标记物 | 检测方法 | 特点 |
| [SB]32[/SB]P | β射线,自显影 | 灵敏度最高,半寿期14天,放射强度1.7MeV |
| [SB]35[/SB]S | β射线,自显影 | 灵敏度高,分辨率好,半寿期87天,0.17MeV |
| [SB]3[/SB]H | β射线,自显影 | 低敏度高,分辨率高,半寿期12年,0.01MeV |
| [SB]125[/SB]I | γ射线,自显影 | 灵敏度高,分辨率高,半寿期60天,0.04MeV |
| 生物素 | 酶标血凝素,显色 | 灵敏度好,避免有内源性生物素标本 |
| 地高辛 | 酶标地高辛配体,显色 | 灵敏度好,分辨率一般 |
| T-T二聚体 | 酶标抗二聚体抗体,显色 | 灵敏度好,紫外照射形成二聚体而标记 |
| BrdU | 酶标抗BrdU抗体,显色 | 灵敏度好,细菌内含质粒复制掺入 |
| 铕-补骨脂素 | 荧光 | 灵敏度一般 |
缺口平移标记法先由DNase Ⅰ在DNA双链上切出随机切口,DNA聚合酶Ⅰ沿缺口水解5′端核苷酸和3′端修复加入被标记核苷酸同时进行,切口平行推移,可均一标记DNA链。(图18-4、5)
缺口平移法快速、简便、成本相对较低、比活性较高、标记均一。主要利用了DNA聚合酶Ⅰ修复DNA的功能,用于大分子DNA标记(>1000bp最好),单链DNA、RNA不能用该法标记。随机引物法具有上述优点,可代替缺口平移法。此外大小、单双DNA均可标记,标记均匀,标记率高,但不能标记环状DNA。利用末端转移酶可进行“尾标”,尾标适用于寡核苷酸探针标记,用于大分子核酸标记因尾巴短而标记比活性降低。尾标不能用dTTP标记,因mRNA的多聚(A)尾而影响杂交特异性。寡核苷酸探针多用于核酸“点”突变检测,该探针可用核酸合成仪人工合成。克隆探针一般较长,特异性
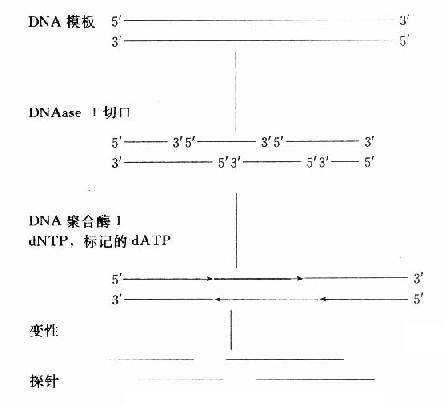
图18-5 DNA随机引物标记法
好,标记量大,杂交检出信号强。合成探针长度短,一般在20-50核苷酸之间,过长合成成本高,且易出现聚合酶合成错误,杂交时间长。太短则特异性下降。合成探针设计还应注意碱基组成G≡C含40%-60%,一种碱基连续重复不超过4个,以免非特异性杂交产生。还要注意探针自身序列内应无互补区域以免产生“发夹”结构,影响杂交。一个好的探针最终在实践中才能加以确认。另外,还有利用M13噬菌体载体可复制单链DNA的特点,复制合成DNA探针,利用转录载体(DNA),转录合成RNA探针。下面就GIBCoBRL公司(Bethesda Research Laboratories美国)的缺口平移法标记生物素试剂盒(bionick labeling system)为例介绍如下:
| 试剂: | 10x dNTP Mix.250μl | 10x Enzyme Mix 250μl |
| 0.2mmol/L each dCTP,dCTP,dTTP | 0.5 units/μl DNA Polymerase Ⅰ | |
| 0.1mmol/L dATP | 0.0075 units/μl DNase Ⅰ | |
| 0.1mmol/L biotin-14-dATP | 50mmol/L Tris-HCl(pH7.5) | |
| 500mmol/L Tris-HCl(pH7.8) | 5mmol/L magnesium acetate | |
| 50mmol/L MgCl[XB]2[/XB] | 1mmol/L β-mercaptoethanol | |
| 100mmol/L β-mercaptoethanol | 0.1mmol/L phenylmethyl- | |
| sulfonyl fluoride | ||
| 100μg/ml nuclease-free BSA | 50%(v/v)glycerol | |
| 100μg/ml nuclease-free BSA | ||
| Control DNA:5μg pBR322 in TE | ||
| stop buffer 300 mM EDTA | ||
| autoclaved H[XB]2[/XB]O |
操作步骤:
将5μl 10x dNTP Mix.;-μl DNA(相当1μg需标记的DNA量);-μl灭菌蒸馏水加至45μl;再加5μl10x Enzyme Mix.依次加入1.5-2ml离心管后。15000xg离心15秒钟。16℃保温1小时。加5μl Stop Buffer终止反应。乙醇沉淀,备用。